
Zespół Stevensa-Johnsona – jakie są objawy i jak wygląda leczenie tej choroby skóry?
min. czytania
Gorączka, wysypka, bolesne nadżerki w jamie ustnej – na pierwszy rzut oka mogą przypominać zwykłą infekcję, ale w rzeczywistości mogą być objawem zespołu Stevensa Johnsona. To choroba, która w krótkim czasie może doprowadzić do rozległych uszkodzeń skóry i narządów. Dlatego, jeśli zauważysz podobne sygnały, nie zwlekaj z reakcją – wczesna pomoc medyczna może uratować zdrowie, a nawet życie. Z tego artykułu dowiesz się, czym jest zespół Stevensa-Johnsona, jak go rozpoznać i na czym polega leczenie.
Spis treści
Zespół Stevensa-Johnsona (SJS) – co to jest?
Zespół Stevensa-Johnsona (SJS) to rzadka, ale bardzo poważna choroba skóry i błon śluzowych. Najczęściej pojawia się jako reakcja na leki lub – rzadziej – w przebiegu infekcji. Choroba zaczyna się nagle i w krótkim czasie może doprowadzić do poważnych uszkodzeń skóry oraz narządów wewnętrznych.
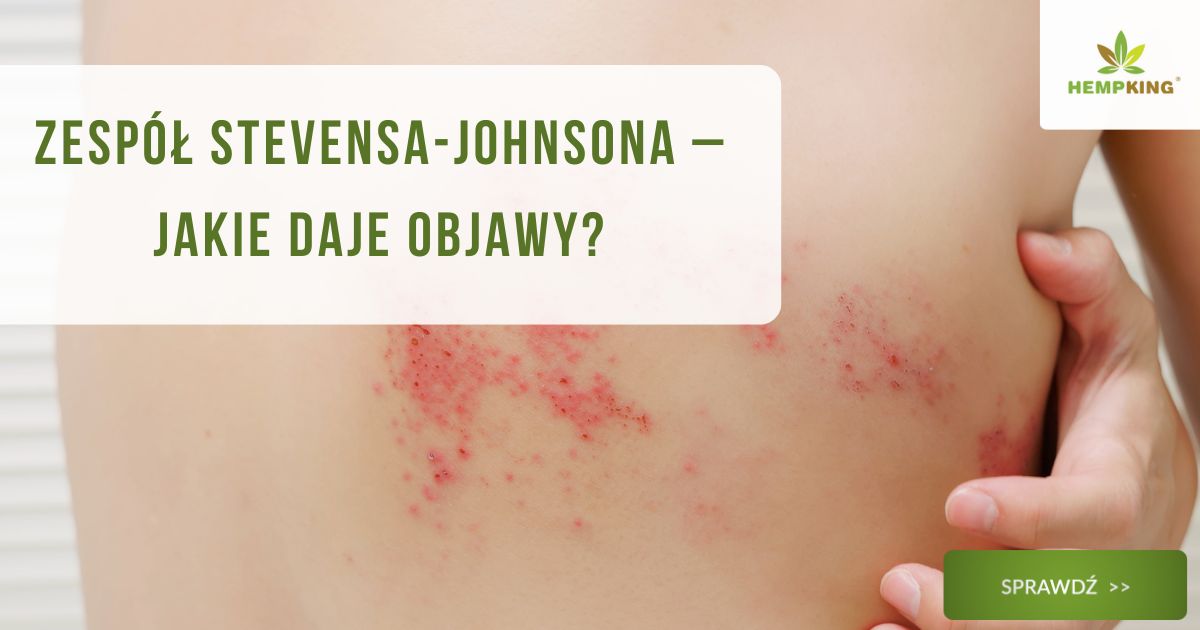
W SJS układ odpornościowy reaguje w sposób nadmierny, powodując martwicę komórek skóry. W efekcie powstają bolesne nadżerki i pęcherze, które przypominają rozległe oparzenia. Zmiany obejmują nie tylko skórę, ale też usta, oczy czy narządy płciowe.
Ta choroba zawsze wymaga leczenia szpitalnego, często na oddziale intensywnej terapii lub w specjalistycznym ośrodku leczenia oparzeń. Wczesne rozpoznanie ma kluczowe znaczenie – im szybciej zostanie przerwane działanie czynnika wywołującego, tym większe szanse na ograniczenie powikłań.
Jakie są przyczyny zespołu Stevensa-Johnsona?
Najczęściej zespół Stevensa-Johnsona rozwija się jako reakcja niepożądana na leki. Organizm traktuje substancję aktywną jak zagrożenie i uruchamia silną odpowiedź immunologiczną. Skutkiem jest uszkodzenie komórek skóry i błon śluzowych.
Poza lekami istnieją także inne możliwe przyczyny. U niektórych osób SJS rozwija się w następstwie infekcji wirusowych lub bakteryjnych. Zdarza się również, że choroba pojawia się bez uchwytnej przyczyny, choć takie przypadki należą do wyjątków.
Warto pamiętać, że nie każdy lek czy infekcja prowadzi do SJS. Ryzyko rośnie, gdy nakłada się kilka czynników – na przykład predyspozycje genetyczne, osłabiony organizm i równoczesne stosowanie kilku różnych preparatów.
Jakie leki wiążą się z największym ryzykiem wystąpienia SJS?
Największe ryzyko pojawienia się zespołu Stevensa-Johnsona dotyczy kilku konkretnych grup leków. W praktyce oznacza to, że jeśli je stosujesz, lekarz powinien monitorować Twój stan i reagować na pierwsze niepokojące objawy.
Do leków najczęściej powiązanych z rozwojem SJS należą:
- antybiotyki sulfonamidowe – dawniej stosowane szerzej, dziś rzadziej, ale nadal mogą wywoływać silne reakcje,
- leki przeciwpadaczkowe – takie jak karbamazepina, fenytoina czy lamotrygina,
- niesteroidowe leki przeciwzapalne (NLPZ) – zwłaszcza z grupy oksykamów, np. piroksykam,
- allopurynol – stosowany w leczeniu dny moczanowej.
Ryzyko jest większe, gdy lek przyjmujesz po raz pierwszy lub gdy dawka została zwiększona. Objawy mogą wystąpić po kilku dniach, ale też dopiero po kilku tygodniach od rozpoczęcia terapii.
Jeśli stosujesz jeden z tych leków i zauważysz wysypkę, ból w ustach czy podrażnienie oczu – nie bagatelizuj tego sygnału i skontaktuj się z lekarzem.
Czy infekcje również mogą być przyczyną SJS?
Tak, choć rzadziej niż leki. U części osób zespół Stevensa-Johnsona pojawia się w następstwie infekcji. Najczęściej są to zakażenia wirusowe, takie jak:
- opryszczka (HSV),
- zapalenie płuc wywołane przez Mycoplasma pneumoniae,
- grypa czy inne infekcje dróg oddechowych.
Zdarza się również, że SJS rozwija się po infekcjach bakteryjnych lub grzybiczych, ale to sytuacje zdecydowanie mniej typowe.
Infekcja może być jedynym czynnikiem wywołującym chorobę albo działać razem z innymi – np. gdy w tym samym czasie przyjmujesz nowy lek. Wtedy ryzyko gwałtownej reakcji jest większe.
Kto znajduje się w grupie podwyższonego ryzyka?
Nie każdy, kto przyjmuje leki czy przechodzi infekcję, zachoruje na zespół Stevensa-Johnsona. Istnieją jednak czynniki, które zwiększają ryzyko.
Do grupy podwyższonego ryzyka należą osoby:
- które wcześniej miały reakcję alergiczną na określony lek,
- z predyspozycjami genetycznymi – u niektórych populacji wykryto określone warianty genów zwiększające ryzyko (np. HLA-B*1502 u osób pochodzenia azjatyckiego),
- stosujące kilka leków jednocześnie, zwłaszcza z grup ryzykownych,
- z osłabionym układem odpornościowym – np. w przebiegu chorób autoimmunologicznych, nowotworów czy zakażenia HIV,
- w młodszym wieku – SJS może wystąpić także u dzieci, zwłaszcza po infekcjach wirusowych.
Jeśli jesteś w grupie ryzyka, warto informować lekarzy o wcześniejszych reakcjach i zachować szczególną czujność przy wprowadzaniu nowych leków.

Jakie są pierwsze objawy zespołu Stevensa-Johnsona?
Pierwsze sygnały SJS często przypominają zwykłą infekcję. To sprawia, że choroba na początku bywa bagatelizowana. Zanim pojawią się zmiany skórne, możesz odczuwać:
- gorączkę,
- ból gardła,
- kaszel,
- bóle mięśni i ogólne osłabienie.
Po kilku dniach pojawia się bolesna wysypka. Najpierw wygląda jak drobne, czerwone plamy, które szybko przekształcają się w pęcherze i nadżerki. W tym samym czasie zaczynają być zajęte błony śluzowe – usta, oczy czy narządy płciowe.
Charakterystyczne jest też to, że zmiany pojawiają się nagle i szybko się rozprzestrzeniają. Towarzyszy im ból, pieczenie i trudności w jedzeniu czy połykaniu.
Gdzie na ciele lokalizują się zmiany w przebiegu SJS?
Zespół Stevensa-Johnsona to choroba skóry i błon śluzowych, w której zmiany nie ograniczają się do jednego miejsca. Już we wczesnym etapie pojawia się rumień, który szybko przekształca się w pęcherze i bolesne nadżerki.
Najczęściej zajęte są:
- twarz i tułów – początkowe ogniska wysypki, które mogą przypominać rumień wielopostaciowy,
- kończyny – zmiany często rozprzestrzeniają się na ręce i nogi,
- błony śluzowe jamy ustnej – bolesne owrzodzenia i nadżerki utrudniają jedzenie i picie,
- narządy płciowe – zmiany na błonach śluzowych w tej okolicy są szczególnie dokuczliwe i mogą prowadzić do powikłań,
- oczy – ich zajęcie uznaje się za jeden z najgroźniejszych objawów zespołu Stevensa-Johnsona.
Charakterystyczne jest to, że zmiany rozwijają się gwałtownie i przypominają oparzoną skórę. W ciężkich przypadkach dochodzi do odwarstwienia naskórka na dużej powierzchni, co wiąże się z ryzykiem odwodnienia, zakażenia i poważnych powikłań ogólnoustrojowych.
Jak wyglądają zmiany na błonach śluzowych?
W zespole Stevensa-Johnsona zmiany na błonach śluzowych są jednym z najbardziej charakterystycznych objawów. Zwykle pojawiają się wcześnie i szybko się nasilają.
Najczęściej dotyczą:
- jamy ustnej – występują bolesne nadżerki i owrzodzenia, które utrudniają jedzenie, picie, a nawet mówienie,
- warg – tworzą się bolesne strupy i pęknięcia,
- narządów płciowych – zmiany powodują silny ból i mogą prowadzić do bliznowacenia,
- błony śluzowej oczu – rozwija się silny stan zapalny, a ropna wydzielina dodatkowo nasila dolegliwości.
Zmiany są rozległe i przypominają powierzchnię oparzenia. Pacjent odczuwa silny ból, a uszkodzona błona śluzowa staje się podatna na zakażenia. Nadżerki i pęcherze mogą się nadkażać, co zwiększa ryzyko powikłań.
Dlaczego SJS jest tak niebezpieczny dla oczu?
Zajęcie oczu w przebiegu zespołu Stevensa-Johnsona to jeden z najpoważniejszych objawów. Zmiany w tej okolicy nie tylko powodują silny ból i pieczenie, ale mogą prowadzić do trwałych uszkodzeń wzroku.
Na początku pojawia się:
- silne zaczerwienienie spojówek,
- obrzęk powiek,
- uczucie piasku pod powiekami i światłowstręt.
W ciężkich przypadkach dochodzi do powstania nadżerek i blizn na rogówce. To właśnie one odpowiadają za pogorszenie, a nawet całkowitą utratę widzenia. U części pacjentów rozwija się przewlekły zespół suchego oka, który wymaga stałego leczenia.
Dlatego każde podejrzenie zajęcia oczu w SJS wymaga natychmiastowej konsultacji okulistycznej. Wczesne włączenie odpowiedniego leczenia może ograniczyć ryzyko nieodwracalnych powikłań.



Jak wygląda diagnostyka i leczenie zespołu Stevensa-Johnsona?
Rozpoznanie zespołu Stevensa-Johnsona opiera się przede wszystkim na obrazie klinicznym. Lekarz ocenia, jak wyglądają zmiany skórne i błon śluzowych oraz zbiera dokładny wywiad dotyczący leków i przebytych infekcji. W niektórych przypadkach wykonuje się badania dodatkowe – np. biopsję skóry – aby potwierdzić diagnozę i odróżnić SJS od toksycznej nekrolizy naskórka (TEN).
Diagnostyka obejmuje:
- analizę przyjmowanych leków i możliwych infekcji,
- badanie zmian skórnych (pęcherze, nadżerki, rumień),
- ocenę zajęcia błon śluzowych jamy ustnej, narządów płciowych i oczu,
- badania krwi, a czasem obrazowe, aby ocenić stan narządów wewnętrznych.
Leczenie zespołu Stevensa-Johnsona zawsze wymaga hospitalizacji – często na oddziale intensywnej terapii lub w ośrodku leczenia oparzeń. Pierwszym krokiem jest natychmiastowe odstawienie leku, który mógł wywołać objawy.
Podstawowe elementy leczenia to:
- intensywna pielęgnacja skóry jak w przypadku oparzeń,
- nawodnienie i uzupełnianie elektrolitów,
- leki przeciwbólowe i przeciwzapalne,
- antybiotyki w razie nadkażeń,
- specjalistyczne leczenie okulistyczne, jeśli zajęte są oczy.
W ciężkich przypadkach stosuje się także leki immunosupresyjne lub dożylne immunoglobuliny, aby zmniejszyć nadmierną reakcję organizmu.
Jakie mogą być długofalowe skutki i powikłania po SJS?
Zespół Stevensa-Johnsona to choroba skóry, która nawet po wyleczeniu może pozostawić trwałe konsekwencje. U wielu pacjentów dochodzi do bliznowacenia skóry i błon śluzowych, a także do zaburzeń pigmentacji, które utrzymują się przez długi czas. Zmiany w obrębie jamy ustnej mogą powodować przewlekły ból i trudności z jedzeniem, a w przypadku zajęcia narządów płciowych mogą prowadzić do powstawania zrostów i dolegliwości bólowych.
Jednym z najgroźniejszych powikłań jest uszkodzenie oczu. Nawet po zakończonym leczeniu choroby ryzyko trwałej utraty ostrości wzroku czy rozwoju zespołu suchego oka pozostaje wysokie. Niekiedy konieczna jest wieloletnia kontrola okulistyczna i stosowanie specjalistycznych preparatów.
SJS może również wpływać na funkcjonowanie narządów wewnętrznych. W ciężkich przypadkach rozwija się ostra niewydolność oddechowa, uszkodzenie nerek lub powikłania w obrębie układu pokarmowego. Te następstwa często wymagają dalszej opieki i rehabilitacji.
Powikłania psychiczne także są częste – pacjenci po przebyciu zespołu Stevensa i Johnsona mierzą się z lękiem przed ponownym zachorowaniem i obawą przed przyjmowaniem nowych leków. Choroba przypomina o sobie przez wiele lat, dlatego niezbędne jest nie tylko leczenie objawów, ale również wsparcie psychologiczne.
Zespół Stevensa-Johnsona nie kończy się w momencie ustąpienia ostrych objawów. Skutki mogą być długofalowe i dotyczyć zarówno zdrowia fizycznego, jak i psychicznego.

Badania nad zespołem Stevensa Johnsona
Badania nad zespołem Stevensa–Johnsona coraz częściej koncentrują się na mechanizmach immunologicznych. Wykazano, że choroba wynika z nadmiernej aktywacji limfocytów T, prowadzącej do apoptozy keratynocytów i martwicy naskórka. Naukowcy analizują rolę cytokin prozapalnych oraz genetycznych czynników ryzyka, co pozwala rozwijać bardziej precyzyjne terapie ukierunkowane na reakcje układu odpornościowego. Dużą uwagę poświęca się także szybszej diagnostyce.
Zespół Stevensa–Johnsona to choroba ogólnoustrojowa, dlatego ocena jej ciężkości wykracza poza analizę zmian skórnych. Skala SCORTEN uwzględnia m.in. wiek, wydolność narządów, choroby współistniejące oraz wyniki badań laboratoryjnych wskazujących na zaburzenia metaboliczne czy niewydolność nerek i układu krążenia. Takie podejście pozwala lepiej ocenić ryzyko powikłań i dobrać właściwe leczenie [1].
Toksyczna nekroliza naskórka najczęściej rozwija się w pierwszych tygodniach po rozpoczęciu nowego leku – zwykle w ciągu miesiąca, choć czasem później. To okres największego ryzyka, dlatego pacjent powinien uważnie obserwować organizm, zwłaszcza jeśli wcześniej reagował źle na leki. Wczesne objawy mogą przypominać infekcję, co utrudnia rozpoznanie, jednak szybkie odstawienie preparatu znacząco zmniejsza ryzyko ciężkiego przebiegu choroby. Dlatego kluczowa jest czujność w początkowej fazie terapii [2].
U osób, które mają być leczone lekami mogącymi wywołać zespół Stevensa-Johnsona, coraz częściej zwraca się uwagę na badania profilaktyczne. Najważniejsze są testy genetyczne, które pozwalają sprawdzić, czy ktoś ma wrodzoną podatność na taką reakcję. W niektórych krajach, takie badania wykonuje się rutynowo, zanim pacjent zacznie przyjmować np. leki na padaczkę czy dnę moczanową. W Europie nie jest to jeszcze powszechna praktyka, ale temat coraz częściej pojawia się w zaleceniach lekarskich. Oprócz testów genetycznych ważne są też regularne badania krwi, szczególnie w pierwszych tygodniach leczenia. Dzięki nim można zauważyć niepokojące zmiany, zanim rozwinie się ciężka choroba [3].
Zespół Stevensa Johnsona | Podsumowanie
Zespół Stevensa-Johnsona to rzadka, ale bardzo poważna choroba skóry i błon śluzowych, która najczęściej rozwija się jako reakcja na lek lub infekcję. Objawy początkowo mogą przypominać zwykłe przeziębienie, jednak szybko pojawiają się rumień, pęcherze i bolesne nadżerki. Choroba zawsze wymaga hospitalizacji i natychmiastowego odstawienia leku, który wywołał objawy. Szybkie rozpoznanie i odpowiednie leczenie są kluczowe, by ograniczyć ryzyko powikłań, takich jak uszkodzenie wzroku czy zajęcie narządów wewnętrznych. Nawet po wyleczeniu pacjent często potrzebuje długoterminowej opieki medycznej.
FAQ
Czym różni się zespół Stevensa-Johnsona od zespołu Lyella?
Zespół Stevensa-Johnsona i zespół Lyella to dwa schorzenia z tej samej grupy ciężkich reakcji skórnych. Różnią się jednak zakresem uszkodzenia skóry. W SJS zmiany obejmują do 10% powierzchni ciała, natomiast zespół Lyella (określany też jako toksyczna nekroliza naskórka) dotyczy ponad 30% powierzchni skóry i ma cięższy przebieg.
Jaka jest śmiertelność w przebiegu zespołu Stevensa-Johnsona?
Śmiertelność w SJS zależy od wielu czynników, m.in. wieku pacjenta, ogólnego stanu zdrowia, zajęcia narządów wewnętrznych oraz czasu, w jakim rozpoczęto leczenie. Szacuje się, że umieralność w SJS wynosi około 5–10%, a w cięższych postaciach, jak toksyczna nekroliza naskórka, może być znacznie wyższa.
Jak długo trwa leczenie zespołu Stevensa-Johnsona?
Czas leczenia zależy od ciężkości przebiegu choroby i ogólnego stanu pacjenta. U części osób hospitalizacja trwa kilka tygodni, ponieważ trzeba zadbać o regenerację skóry i błon śluzowych oraz kontrolować możliwe powikłania. Nawet po wyjściu ze szpitala konieczna jest długofalowa opieka, np. okulistyczna lub dermatologiczna.
Zobacz także
Od czego są zależne choroby skóry?
Choroby włosów i skóry głowy – jak je diagnozować i skutecznie leczyć?
Tłuszczak – przyczyny, diagnoza i metody leczenia
Bibliografia
- Hasegawa A, Abe R. Recent advances in managing and understanding Stevens-Johnson syndrome and toxic epidermal necrolysis. F1000Res. 2020 Jun 16;9:F1000 Faculty Rev-612.
- Shah H, Parisi R, Mukherjee E, Phillips EJ, Dodiuk-Gad RP. Update on Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: Diagnosis and Management. Am J Clin Dermatol. 2024 Nov;25(6):891-908.
- Liotti L, Caimmi S, Bottau P, Bernardini R, Cardinale F, Saretta F, Mori F, Crisafulli G, Franceschini F, Caffarelli C. Clinical features, outcomes and treatment in children with drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis. Acta Biomed. 2019 Jan 29;90(3-S):52-60.















Dodaj komentarz